
- Needs a bit on the various loop diuretics
- Needs the data showing harm from diuretics with ADHF
- Updated 4/18/11
- 82 slides, 1:22 minutes


musings of a salt whisperer

Sodium including electrolyte free water (Keynote, Powerpoint PDF)
This is an older version of my hyponatremia lecture. This lecture has gone through various versions and though I like my most current version, others may like older variations better. Additionally the Keynote version (2014) is similar but not identical to the Powerpoint version (2013).

Hi Dr. Topf
First of all, apologies for sending this via email but I do not have a Twitter account (I know, its the 21st century, who doesn’t?).
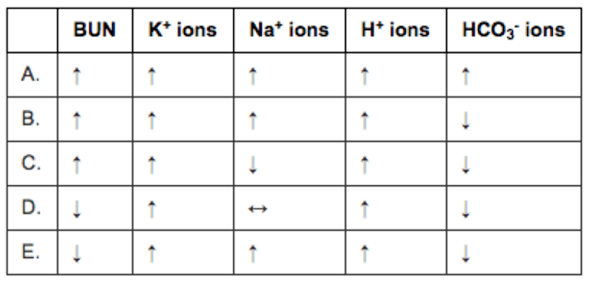
I had a quick question regarding a practice problem I was doing. Rather than summarize the question for you, I included a screenshot so that you have the primary source with the explanation provided. Below, I also included my explanation for my reasoning for choosing that option. Basically, I am confused as to why the bicarb would be decreased in this scenario.
So the stem describes acute trauma. Specifically crush injuries, so you should be thinking rhabdomyolysis where the body gets turned inside out. In my very first lecture we talked about the intracellular atmosphere versus the extracellular atmosphere:



I posted this to twitter. The subsequent discussion was pretty interesting:

Hello Dr. Topf,
I hope you are enjoying your weekend. I had a question in regards to one of your lectures. I was wondering why there is a low level of Uric acid in euvolemic hyponatremia but not in hypervolemic or hypovolemic hyponatremia. Also, how is it that Na taken in equals Na excreted in euvolemic hyponatremia?
All the best,


This is similar to what we see with urea. The following description of urea handling gives a model that will work for uric acid, though the truth of uric handling is much more complex.
Hi Dr. Topf,
I had a question regarding the Macula Densa. When reviewing your powerpoint on volume control, you have a slide that said there is only one osmoreceptor (Hypothalamus) because osmolarity across the body is the same at all times. I’ve had some confusion regarding the Macula Densa, but from what I understand it is also an osmoreceptor (sensing Na+ in the tubule), which would make sense because the tubules are the only part of the body where osmolarity is different.
I thought that the Macula Densa would affect GFR and stimulate the release of renin from the Juxtaglomerular cells, but that would seem to affect volume (RAAS System maintains volume), so my question is why does the macula densa (which senses Na+) controlling volume and not Osmolarity?
Good question.
So the macula densa is a major part of a process called tubulo-glomerular feedback
As the name implies this is important for balancing GFR with tubular reabsorption.
If you had excess GFR and limited tubular reabsorption, people could literally pee them selves to death in minutes.
Think about the math, you have 3 liters of plasma and filter 125 ml of it every minute. so it would only take 24 minutes to completely filter all of the plasma. if you are not constantly reabsorbing 99% of the filtered fluid you could very rapidly become volume depleted and suffer from cardiovascular collapse.
Tubular glomerular feedback prevents that. At the end of the thick ascending limb of the loop of henle, there are chloride receptors as part of the juxtaglomerular apparatus. If there is too much filtration and not enough reabsorption, the excess chloride will bind these receptors and cause a release of intra-renal signals that decrease GFR by adjusting the dilation of the afferent and efferent arterioles.
So yes there are receptors that bind chloride and you can think of them responding to the various concentrations of chloride (like an osmoreceptor) but they are not involved in volume regulation or osmoregulation, but rather the safe running of the kidney to prevent a person from accidentally peeing themselves to death.
Hope this helps
Hello Dr. Topf,
I hope you are doing well. I had a few questions in regard to your last lecture at OUWB. I was wondering if you could explain the pathophysiology behind euvolemic hyponatremia caused by hypothyroidism and adrenal insufficiency. Also, in the case of SIADH, whay wouldn’t the person have hypervolemia if there is a constant reabsorption of water? Is there a pathophysiologic explaination for this as well? I could not find any answers online.
So the key here is to remember that volume is determined by total body sodium and that SIADH is generally Na in = Na out. So they are in sodium balance and will not be volume overloaded.
You are right that these patients will have excess water, but much of this water disappears into the intracellular compartment and the excess volume can not be picked up clinically (by exam or by conventional blood and radiology tests). Yes there is excess water.
We try to reserve terms like hypervolemia for excess total body sodium, and this is not found in SIADH.
Hope that is helpful.

For the sixth year I have had the privledge of teaching at OUWB. When I teach I get e-mail questions from the students. I respond to the students by e-mail but also post the questins and answers here so all the students get the advantages of the questions.




 There is some ambiguity on whether glucose induced hyponatremia can be called pseudohyponatremia, there is some support for it and I used to be in that camp (in fact I wrote a whole book about fluid electrolytes waving the glucose induced pseudohyponatremia flag) but most people limit the term pseudohyponatremia to just the high protein and high lipids causing the lab error (sometimes called a lab artifact), and separate out the high osmolar causes under a different category (factitious hyponatremia).
There is some ambiguity on whether glucose induced hyponatremia can be called pseudohyponatremia, there is some support for it and I used to be in that camp (in fact I wrote a whole book about fluid electrolytes waving the glucose induced pseudohyponatremia flag) but most people limit the term pseudohyponatremia to just the high protein and high lipids causing the lab error (sometimes called a lab artifact), and separate out the high osmolar causes under a different category (factitious hyponatremia).An e-mail I received last month:
I love your blog. I have had SIADH for a zillion years. I only found out what I had when I went with 5 girlfriends to a fancy spa hotel in Tucson for a mini-vacation/ 4th of July Weekend in 1997 where the heat increased to an uncomfortable 117 degrees.
Healthcare workers in the hotel kept handing out bottles of water at each hotel exercise location with orders to “hydrate, hydrate, hydrate” and I stupidly followed their directions. I drank myself into a 6 day coma.
 |
| The only time that sentence has been used for water, not alcohol. |
Very non-traumatic for me. Very traumatic for my family. I woke up on day 6 saying, “I am STARVING! Will someone go get me a taco?” which was very anxiety-relieving for all of them; they’d been sure I’d wake up cognitively impaired. I wasn’t. This “taco” sentence sounded JUST like me. And I have continued to be not cognitively impaired despite interesting lab numbers.
My dad (who is a physician too) has SIADH as well, though his was diagnosed after mine. I was mis-diagnosed for 9 years prior to my coma as having a “seizure disorder.” The excellent care I received when my mental status went to heck in a handbasket was truly life-saving. I remain a very grateful nephrology patient. And I really do love your blog.
Last year I had one of the most severe cases of hyponatremia I had ever seen. Last night I was flipping through iPhoto (looking for the drawings Margret Atwood did of my alter ego, Kidney Boy) and found this photo…

Just looking at the labs gives me chills. Here is the whole story.
Almost all of the hyponatremia I see is inpatient, but this week a woman was referred to my clinic with a sodium of 128. She has a sharp family doctor who ordered all the right tests. Here are the key pieces:
