
So drop box behaves strangely with Keynote files. When you get to this page by clicking the above links, press the Download button.

Here is a link to the Keynote presentation exported to powerpoint.


musings of a salt whisperer
So drop box behaves strangely with Keynote files. When you get to this page by clicking the above links, press the Download button.
Here is a link to the Keynote presentation exported to powerpoint.

I recently saw a patient with hyponatremia and cirrhosis. He had been started on salt tablets to try to correct the hyponatremia. In a limited number of diagnosis salt tablets can help hyponatremia. Cirrhosis is not one of them. I created this Tweetorial to teach some basic hyponatremia physiology and clear up when they may work and when they will not.
I’m going to throw my hat in the Tweetorial ring.
Sometimes salt tablets work for hyponatremia and sometimes they don’t. How can you figure out when to use them and when not to?
— Joel M. Topf, MD FACP (@kidney_boy) July 9, 2018
This is my first shot at a Tweetorial. I wrote out the tweets in Apple Notes. I think I will use a spreadsheet with a character counter next time. Too many of my Tweet-length thoughts ended up being a bit too wordy. Having a counter during creation would help.
I also should have more links and pics.
Additionally, I bought the domain Tweetorial.org. Any ideas what I should use it for?
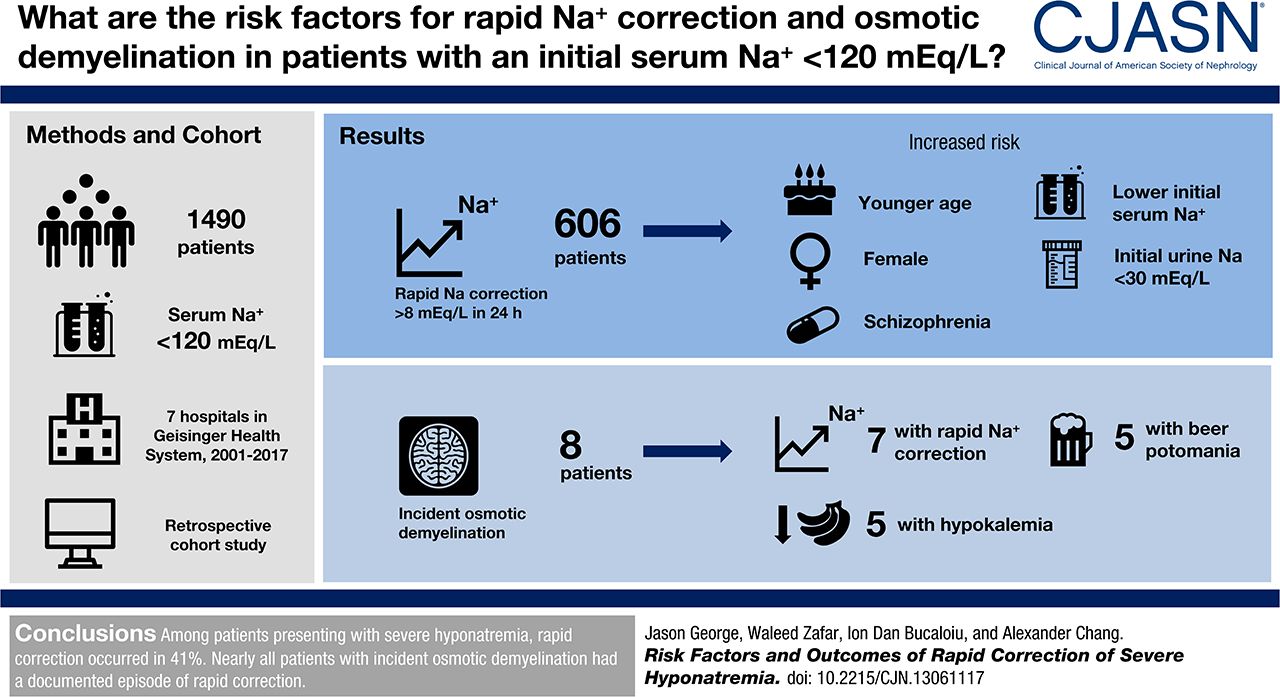 I think I may have buried the lead in the last blog post. Perhaps this imaginary conversation will clarify what is so important about the George et al study.
I think I may have buried the lead in the last blog post. Perhaps this imaginary conversation will clarify what is so important about the George et al study.
Nephrologist: There are now clinical practice guidelines, based on expert opinion, that tell us to correct sodium no more than 8 mmol/L in the first day of hyponatremia.
Internist: That seems cautious. How well are we meeting those guidelines?
Nephrologist: Well in a recent multi-center, retrospective analysis, of nearly 1500 people with an initial sodium below 120, just over 40% went too fast.
Internist: FORTY PERCENT were too fast. Oh my god! We’re doing horrible!
Nephrologist: Yeah, it’s kind of embarrassing.
Internist: So what happened to the 600+ people where the speed limit was exceeded?
Nephrologist: Well, they found 9 people had osmotic demyelination on MRI. But none of them had any documented permanent neurologic deficits and none of them was diagnosed with central pontine myelinolysis.
Internist: So how many people could we help if we worked on protocols, education and training to get that 40% closer to 5 or 10%?
Nephrologist: Well, since with a 40% miss rate we couldn’t find any harm, I guess we couldn’t expect much improvement with a 5 to 10% miss rate.
Internist: We should probably put our energy elsewhere.
This second article in the hyponatremia data dump comes from CJASN and like yesterday’s post, attempts to clarify who develops rapid correction of hyponatremia.
The U.S. Hyponatremia Guidelines (Verbalis, et al) recommend a speed limit for correcting the sodium of no more than 8 mmol/L in any 24 hour period for patients at high risk of osmotic demyelination (ODS). High risk is defined as anyone with:
One of the items I hit home on in my lectures on hyponatremia is how rare osmotic demyelination actually is. I came to this conclusion when NephJC discussed The European guidelines on hyponatremia. The guidelines review every published case or case series of ODS from 1997 until 2012. Over those 15 years they found 53 cases world wide (See Appendix 6, Sheet 13a). Every year, in the US alone, there are around a million people who develop severe hyponatremia (Na < 126). With that type of denominator, 53 cases seems laughably small.

I have never seen a case of osmotic demyelination syndrome. I have seen a lot of mismanaged hyponatremia.
Never have so many, done so much, to avoid a complication seen by so few.
George, Zafar, Bucaloiu, and Chang analyzed the 7 hospitals in the Geisinger Health System to look at the incidence and risk factors for rapid sodium correction and osmotic demyelination.
They included adults with an initial sodium less than 120 mmol/L.
Their primary endpoint was an increase in sodium of more than 8 mmol/L in the first 24 hours.
Looking at the methods, it was not clear to me how they dealt with patients with multiple admissions for hyponatremia. Were they included multiple times? Did they only include the first admission?
They manually looked through every MRI report that was done after the index sodium to look for reports of ODS.
They found 1,490 cases of severe hyponatremia with complete data that matched their inclusion criteria.
Median change in sodium in the first 24 hours was 6.8 mmol/L.
606 (41%) patients broke the speed limit of 8 mmol/L in the first 24 hours.
The authors provided a lot of depth on the risk factors for rapid correction. Here is the unadjusted risk factors.
Demographic:
Medical history:
Clinical data:
They used three different models for multivariate analysis to generate odds ratios for rapid correction of hyponatremia.

Some comments about the risk factors. I suspect the reason that depression actually represents SSRI induced SIADH that rapidly corrects when the drug is withdrawn. The schizophrenia association may represent psychogenic polydipsia, a common manifestation of schizophrenia. Water restriction will result in prompt normalization of the sodium.
Liver, heart, and cancer are all associated with difficult to treat hyponatremia, so no surprise that they saw lower rates of rapid correction among people with those diseases.
The lower BMI and female gender association with rapid correction may be simply due to lower total body water making these patients more susceptible to rapid changes in serum sodium.
Lower urine sodium may indicate patients with volume depletion hyponatremia, a population predisposed to rapid correction of hyponatremia as soon as the volume depletion is corrected, releasing the hypothalamus from the volume stimulated release of ADH and resulting in rapid drop in ADH and rapid, uncontrolled correction of hyponatremia.

The lower urine osmolality likely represents patients with psychogenic polydipsia, tea and toast syndrome, or patients who are already in the midst of spontaneously correcting their sodiums, all patients who will rapidly correct their sodium.
The people who are seizing are a cohort with, presumably, acute symptomatic hyponatremia and rapid correction is less problematic.
There were 295 patients who had an MRI after having severe hyponatremia. Nine had radiologic evidence of osmotic demyelination. No patients had an ICD diagnosis of central pontine myelinolysis. One patient had MRI evidence of ODS prior to correction of the sodium. Consistent with medical consensus, nearly all of the patents with incident ODS had rapid correction of hyponatremia:
Of the eight (0.5%) patients who developed incident osmotic demyelination, seven (88%) had documented sodium correction > 8 mEq/L during any 24-hour period before brain MRI.
The one patient with evidence of ODS without rapid correction of hyponatremia had been admitted within a month with a sodium of 105. So it is possible the ODS occurred with that episode rather than the one captured in the study.
The authors listed the characteristics of the patients with ODS:
Having hypovolemia in 75% and beer potomania in 63% means that some patients had to have both of these diagnosis. This portrays a misunderstanding of what constitutes beer potomania. Beer potomania induced hyponatremia is not merely a patient who drinks beer and develops hyponatremia, instead it represents a patient who is unable to clear the water volume in beer due to inadequate solute intake. These patients have maximally dilute urine. On the other hand, patients with volume induced hyponatremia are unable to clear excess water because of hypovolemia-induced ADH release. In one case you have high ADH activity and concentrated urine (volume depletion) and in the other case you have suppression of ADH and dilute urine. There can not be a meaningful combination of the two disorders.
Alcohol use, malnutrition and hypokalemia are all previously recognized risk factors for ODS.
Three patients had received 3% saline for acute neurologic symptoms of hyponatremia. This is critical information because it shows that just because someone has acute manifestations of hyponatremia and an indication for 3%, one must still pay attention to the speed limit. Often acute neurologic symptoms of hyponatremia is thought to indicate a failure to have compensated for chronic hyponatremia and therefore protective against ODS. This is not the case.
The finding that I found most interesting was the clinical outcomes of the patients with ODS.
Five patients with documented osmotic demyelination had recovery with no neurologic deficits, two patients died from unrelated causes, and two were lost to follow-up.
They had 9 cases of ODS and they weren’t able to document even a single case of neurologic devastation. Five of the cases had documented neurologic recovery. This is how Burton Rose describes ODS:

And here is how Sarah and I described it in the fluids book

In my mind if you don’t spasmodic, mirthless laughter you don’t have ODS.
This is consistent with a previous study of dialysis patients with hyponatremia who developed similarly clinically invisible ODS was diagnosed by MRI. From the discussion of Tarhan et al.
Contrary to its classic description, osmotic demyelination syndrome may develop without any presenting signs and symptoms in patients with end-stage renal disease who have undergone recent hemodialysis.
This largely asymptomatic ODS likely explains why not one of the 9 patients had an ICD diagnosis of central pontine myelinolysis (CPM). They did nt have any of the clinical manifestations of CPM.
When I read the abstract and saw that they had 9 cases of ODS in their cohort I jumed out of my shorts. This would be the largest series of DS ever. After reading the study I realize that actually we are in a new era of hyponatrmia where ODS can be devastating (read the three case reports in Brunner et al’s prospective study of MRI evaluation, PDF available on SciHub) or asymptomatic.
In the last couple of months there has been an outpouring of new hyponatremia data and resources. The first I want to discuss is data on the speed of sodium correction with tolvaptan.
 Juan Carlos lead a group who looked at the speed of sodium rise with tolvaptan. The primary endpoint was the change in sodium at 24-hours in patients given 15 mg of tolvaptan.
Juan Carlos lead a group who looked at the speed of sodium rise with tolvaptan. The primary endpoint was the change in sodium at 24-hours in patients given 15 mg of tolvaptan.
All patients had to have failed fluid restriction to be included in the analysis.
For the purpose of the study, SIADH was defined as:
CHF induced hyponatremia was defined as:
All patients had to start with a tolvaptan dose of 15 mg.
The only other concurrent therapy allowed was fluid restriction. Patients who subsequently were started on D5 water, diuretics or salt tablets, had their data censored at the point where the additional therapies were added.
Diuretics are listed as an exclusion criteria but the CHF group were allowed to use them (an exclusion to the exclusion criteria). This is not well described in the methods.


After restricting the patients by their pre-specified exclusion criteria they had 28 patients with SIADH and 39 with CHF.
Table 1.

Remember how the urine sodium is supposed to be low in heart failure. Take a look at the elevated level found in this study. Conclusion: diuretics work. Also take a look at the low Bun and low uric acid in the SIADH group. These are really helpful in my experience at differentiating the cause in tricky cases.
Tolvaptan was much more effective in SIADH with an average change sodium of 0.80 mmol/L/hr versus 0.17 mmol/L/hr in CHF
Sodium went up by more than 12 mEq/L in 25% of patients with SIADH and 3% of patients with CHF.
Using linear mixed-effects models to conduct multivariable repeated-measures analysis the investigators found:
This is what these variables look like when mixed together (data for SIADH patients)

The discussion includes this tidbit where the investigators try to explain why there is a more dramatic response in SIADH than in CHF.
As seen in Table 1, average kidney function of patients with SIADH was significantly greater than that of patients with CHF. As shown in Figures 4 and 5, a total of 8 of 39 patients with CHF and 1 of 28 patients with SIADH had serum creatinine concentrations > 1.5 mg/dL. Thus, difference in kidney function may account for the observed difference in therapeutic response between the SIADH and CHF groups.
I don’t find this argument convincing because kidney function was tested to see if it predicted response and though eGFR did correlate with response to tolvaptan in SIADH, it was not an independent predictor of response and was not a predictor of response at all in CHF.
In the SIADH cohort, age and baseline values for serum sodium, serum osmolality, SUN, serum creatinine, MDRD, and CKD-EPI significantly correlated with the magnitude of increase in serum sodium concentration during the first 24 hours of therapy. Unlike those parameters, no significant correlation was found between the initial 24-hour increase in serum sodium concentration and either body weight, body mass index, or baseline urine sodium, urine osmolality, serum uric acid, or serum potassium value. In the CHF cohort, baseline serum sodium, serum osmolality, SUN, serum creatinine, and serum potassium values significantly correlated with the 24-hour increase in serum sodium concentration. Conversely, no significant correlation was found between the initial 24-hour increase in serum sodium concentration and either age, body weight, body mass index, or baseline urine sodium, urine osmolality, MDRD, and CKD-EPI values.
 This article is accompanied by an editorial by NephMadness Selection Committee member Richard Sterns. He does a nice job describing why this rapid increase in sodium in SIADH show in Morris’ paper was not also seen in Schrier’s SALT 1 and 2 paper. In that phase 3 trial that lead to the approval of tolvaptan, there were 51 patients patients with SIADH, and only 3 of them corrected too fast. This is 6%, well below the 25% found in Morris’ study. Sterns points out the relatively high sodiums found in SALT study (no one below 120 and only 30 had a sodium below 130) as a likely explanation.
This article is accompanied by an editorial by NephMadness Selection Committee member Richard Sterns. He does a nice job describing why this rapid increase in sodium in SIADH show in Morris’ paper was not also seen in Schrier’s SALT 1 and 2 paper. In that phase 3 trial that lead to the approval of tolvaptan, there were 51 patients patients with SIADH, and only 3 of them corrected too fast. This is 6%, well below the 25% found in Morris’ study. Sterns points out the relatively high sodiums found in SALT study (no one below 120 and only 30 had a sodium below 130) as a likely explanation.
Sterns wraps up his editorial with a neat description of the pharmacokinetics of tolvaptan and arguing for dosing the drug at 3.75 mg and then repeating the dose as needed every 6 hours to titrate the change in the sodium level. Clever.
The minimally effective tolvaptan plasma concentration to increase urine output is approx. 25 ng/mL, and maximal increases in output occur when tolvaptan concentrations exceed 100 ng/mL. Levels > 25 ng/mL are achieved by doses as low as 3.75 mg, but do not remain at this level for long because the half-life for this dose is a little more than 4 hours. A 15-mg dose achieves peak plasma concentra- tions well above 100 ng/mL in patients with SIADH, enough to sustain a maximum water diuresis for more than 4 hours. A maximum water diuresis can increase the serum sodium concentration by >2.5 mEq/L per hour, yet it is not clear why this would be desired.
The standard practice in the United States is to administer 15 mg of tolvaptan and then encourage water intake to offset the resulting variable (and often large) water losses. Considering the high price of the drug in the United States (w$300 per tablet), this practice is basically flushing money down the toilet…
…A much more desirable outcome in patients with severe hyponatremia would be a modest but sustained increase in urine volume with a resulting slow steady increase in serum sodium concentration. If urine volumes were less massive, free-water restriction could be continued to avoid unwanted exacerbation of hypona- tremia. Theoretically, the desired response could be achieved with initial doses of 3.75 mg, repeating or increasing the dose every 6 hours if necessary, based on results of urine output and/or serum sodium levels measured before each dose, until the target increase in serum sodium level for the day is achieved.
Another source of additional insight on the study is an interview by Tim Yau of Juan Carlos at AJKDbog.org.
Every intern knows that the evaluation of hyponatremia includes a TSH and a cortisol level to rule out hypothyroidism and adrenal insufficiency as occult causes of euvolemic hyponatremia.
The mechanism of adrenal insufficiency is a bit confusing with some sources stating that these patients are volume depleted while others are euvolemic.
@askrenal in regards to hypoNa ep w/@kidney_boy a listener wants to know if adrenal insufficiency causes euvolemic or hypovolemic hyponatremia. Apparently various sources disagree. pic.twitter.com/PKsVqq5LVq
— Matthew Watto (@DoctorWatto) April 9, 2018
In some patients without aldosterone, the patients develop severe salt wasting, become hypotensive and get a non-osmotic release of ADH resulting in hyponatremia. These patients will respond to saline. Treat the hypovolemia and the sodium will go up.
Hyponatremia is a common manifestation of adrenal insufficiency even in cases without adrenal crisis. Giving saline to these patients is not effective at correcting the hyponatremia. Giving cortisol, however, results in a brisk water diuresis and rapid correction of the serum sodium (Oelkers, NEJM, 1989).
In addition to being resistant to saline, ADH antagonists (think tolvaptan) protect against this type of adrenal insufficiency-induced hyponatremia.
This means cortisol corrects an abnormality that is due to excess ADH.
Here is the explanation from the late 90’s from The Fluid, Electrolyte and Acid-Base Companion. 
Bartter and Schwartz original definition of SIADH required a normal cortisol specifically to exclude patients with hypopituitism and primary adrenal insufficiency. In primary adrenal insufficiency, in addition to loss of cortisol there is an aldosterone deficiency which can result in sodium wasting, volume depletion and a non-osmotic (decreased perfusion in this case) stimulates for the release of ADH. In this scenario, the patient should appear salt/volume depleted and would not be considered euvolemic.
A nice review of secondary adrenal insufficiency and hyponatremia was done by Sven Diederich.
Remember:
In Diederich’s review they pulled 139 cases of hyponatremia that were referred to endocrinology. (Clearly this study suffers from profound selection bias. Here is a a cleaner study on the epidemiology of hyponatremia by Schrier, from back in the day.) They found 28 cases of hypopituitism leading to hyponatremia. Patients tended to be older (average age 68) and more female (75%). In 25 cases the hypopituitism had not previous been diagnosed and 12 patients had previously been admitted (between 1 and 4 times) for severe hyponatremia without an adequate diagnosis.
Basal cortisol levels were as follows:
Imaging results:
One had secondary adrenal insufficiency due to chronic treatment with prednisolone because of ankylosing spondylitis
Don’t be the doctor that corrects the hyponatremia but fils to diagnose adrenal insufficiency and discharges and sends the patient home only to redevelop hyponatremia another day. Turn over every stone, especially in patients with SIADH of undetermined etiology.
David Goldfarb (@weddellite) and I put on a fluid, electrolyte and acid-base variety show at KidneyCON. The itinerary:
Both of us had a bunch of additional cases that we didn’t get to so be sure to go deep into the presentation files.
Here are the presentation files:
.@kidney_boy at the fluids and electrolytes workshop #KidneyCON pic.twitter.com/VpGbSc8V05
— Matthew Sparks, MD (@Nephro_Sparks) April 7, 2018
@weddellite and @kidney_boy giving a master class on electrolytes and acid base at #KIDNEYcon pic.twitter.com/xJm4Z2wPaK
— Anna Burgner MD MEHP (@anna_burgner) April 7, 2018
Making D5W and 0.9% NS with @kidney_boy in the #KidneyCON fluid workshop pic.twitter.com/zp9e6AXT8I
— Timothy Yau (@Maximal_Change) April 7, 2018
Make #KIDNEYcon great again? @kidney_boy @weddellite pic.twitter.com/Jleke2ZFKd
— Diana Mahbod, MD, CPE, FASN, FNKF (@DiMiRenalMD) April 7, 2018
@kidney_boy tries to induce hypernatremia and hyperglycemia #KIDNEYCon pic.twitter.com/7EfQsVYxEb
— David S Goldfarb, MD (@weddellite) April 6, 2018
Chef @kidney_boy mixing NS and D5W for administration to #kidneycon attendees pic.twitter.com/ChzftumYeZ
— David S Goldfarb, MD (@weddellite) April 6, 2018
At many hospitals 3% is restricted to central lines or ICUs. This is despite data showing that it is safe enough to use peripherally.
From tonight’s NephJC:
My hospitalists and nursing colleagues freak out about 3% saline. Requires central line and step-down unit bed. #NephJC
— KatieOverV (@KatieKwonMD) March 28, 2018
I believe there is evidence that 3% an be given peripherally. have been considering doing a mythbusting blog on this. #NephJC
— 𝙟𝙤𝙨𝙝 𝙛𝙖𝙧𝙠𝙖𝙨 (he/him) 💊 (@PulmCrit) March 28, 2018
we give sodium bicarb peripherally all the time and that has a Na concentration of 1,000 compared to 513 with 3% #NephJC
— Joel M. Topf, MD FACP (@kidney_boy) March 28, 2018
We give it peripherally too #NephJC
— ChristosArgyropoulos MD, PhD FlozinatorInChief (@ChristosArgyrop) March 28, 2018
There: https://t.co/iAuf9d1x84
— Helbert Rondon, MD, MS, FACP, FASN, FNKF (@NephroMD) March 28, 2018

— Helbert Rondon, MD, MS, FACP, FASN, FNKF (@NephroMD) March 28, 2018

And one more

This is my Water and Osmoles lecture edited for students, specifically pre-clinical M2s.

I then broke this down into two PowerPoint presentations


 W
W