Hi Dr. Topf
First of all, apologies for sending this via email but I do not have a Twitter account (I know, its the 21st century, who doesn’t?).
I had a quick question regarding a practice problem I was doing. Rather than summarize the question for you, I included a screenshot so that you have the primary source with the explanation provided. Below, I also included my explanation for my reasoning for choosing that option. Basically, I am confused as to why the bicarb would be decreased in this scenario.
So the stem describes acute trauma. Specifically crush injuries, so you should be thinking rhabdomyolysis where the body gets turned inside out. In my very first lecture we talked about the intracellular atmosphere versus the extracellular atmosphere:
So expect increased potassium and phosphorus.
The vital signs show a patient with circulatory insufficiency, i.e. shock. There are some initial labs drawn from the blood and urine right before resuscitation is initiated. The urine shows an osmolality of 800 mmol/kg H2O (highly concentrated, indicating a lot of ADH activity) and a urine sodium of 5 mEq/L (very low, indicating increased activity of the renin angiotensin aldosterone system).
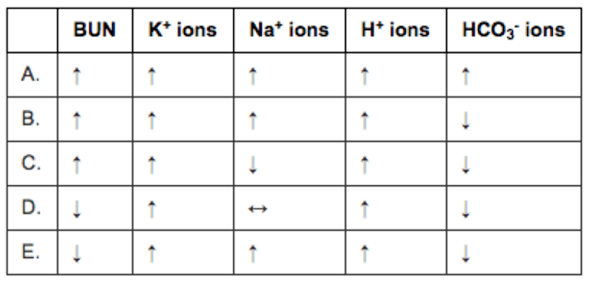
The question then asks you to predict the serum labs. Cool question. The best testing strategy here is to cross off ones that make no sense. Here are the foils:
BUN. This should be elevated as the patient moves to a pre-renal physiology. This leads to an increased filtration fraction to maintain GFR in the face of decreased renal plasma flow. This causes an increase in the osmolality in the efferent arteriole and vasa recta. this increases osmotic reabsorption of fluid from the proximal tubule. BUN flows passively with the fluid, decreasing renal urea clearance and increasing serum BUN. So D is wrong. E is wrong.
Potassium (K+) ions. All the choices show that it rises as the rhabdomyolysis from the crush injury releases loads of intracellular potassium. No answers are eliminated here.
Sodium (Na+) ions. We have a mishmash of choices here. This is difficult to predict. The increased ADH released due to shock would tend to lower the sodium. Increased aldosterone and renin would tend to increase the sodium. Since both are happening together I would expect them to balance each other out resulting in no change in sodium concentration. This is especially true since the stem specifically says there has not been much urine output I would go with D, for no change in sodium concentration. NOTE: About activation of the RAAS as a cause of hypernatremia. Hypernatremia is commonly listed as a symptom of Cushing syndrome and hyperaldosteronism so it is possible to have hypernatremia from (at least the pathological) activation of the renin angiotensin aldosterone system) but this is very unusual as increased in sodium concentration stimulates thirst which dilutes the sodium back to normal.
THIS COLUMN IS MESSED UP. THIS IS AN ERROR BY THE QUESTION AUTHOR.
Hydrogen (H+) ions. All the choices show that it rises. In shock we expect an increase in hydrogen ions as patients move to anaerobic metabolism due to inadequate perfusion (the functional definition of shock). No answers are eliminated here.
Bicarbonate (HCO3-) ions. Bicarbonate is the primary buffer in the body. Increases in hydrogen ions will be buffered by bicarbonate and bicarbonate will be consumed.
So the right answer is bicarbonate will be decreased. This eliminates answer A.
This leaves us with B or C and the question hangs on what the sodium will do and the reality is the change in sodium is unknowable. Shit question. Sorry.
Here is what Kaplan claims to be true:
The forth bullet point is the one where the question fails. It is true that there is accumulation of plasma electrolytes in acute kidney injury. The problem is that these are electrolytes are measured as concentrations and there is also an accumulation of water (which is normally excreted by the kidney). This means that the effect on concentration is variable. Some, like hydrogen and potassium, reliably increase in AKI, but sodium is often decreased in AKI. Maybe the question writers were looking at an unconventional way measuring ions in the the plasma (as total amount rather than concentration).
The line in the answer that pre-renal azotemia is associated with hypernatremia is just wrong. You will encounter numerous patients with pre-renal disease that will have simultaneous hyponatremia. It is impossible to predict the serum sodium concentration from the volume status. This question reinforces the worst instincts of med students when it comes to predicting serum sodium. As I emphasized in the lecture, volume regulation and osmoregulation have two different regulatory systems and, though there is some cross talk, they are largely independent from each other.
The last paragraph tries to make the case that hyponatremia is more common in ATN while hypernatremia predominates in pre-azotemia. This is total fiction and does not exist. Though there can be more urinary sodium in ATN, if the patient is oliguric, it doesn’t matter how high the urine sodium concentration is, if the urine volume volume is close to zero there will not be much urinary sodium excretion.
Distinguishing between pre-renal azotemia and ATN is a constant problem in nephrology. Trust me you can’t make the determination by looking at the serum sodium. It aint that easy.
This question writer should never write another question about sodium.