This second article in the hyponatremia data dump comes from CJASN and like yesterday’s post, attempts to clarify who develops rapid correction of hyponatremia.
The U.S. Hyponatremia Guidelines (Verbalis, et al) recommend a speed limit for correcting the sodium of no more than 8 mmol/L in any 24 hour period for patients at high risk of osmotic demyelination (ODS). High risk is defined as anyone with:
- Na < 105 mmol/L
- Hypokalemia
- Malnutrition
- Liver disease
One of the items I hit home on in my lectures on hyponatremia is how rare osmotic demyelination actually is. I came to this conclusion when NephJC discussed The European guidelines on hyponatremia. The guidelines review every published case or case series of ODS from 1997 until 2012. Over those 15 years they found 53 cases world wide (See Appendix 6, Sheet 13a). Every year, in the US alone, there are around a million people who develop severe hyponatremia (Na < 126). With that type of denominator, 53 cases seems laughably small.

I have never seen a case of osmotic demyelination syndrome. I have seen a lot of mismanaged hyponatremia.
Never have so many, done so much, to avoid a complication seen by so few.
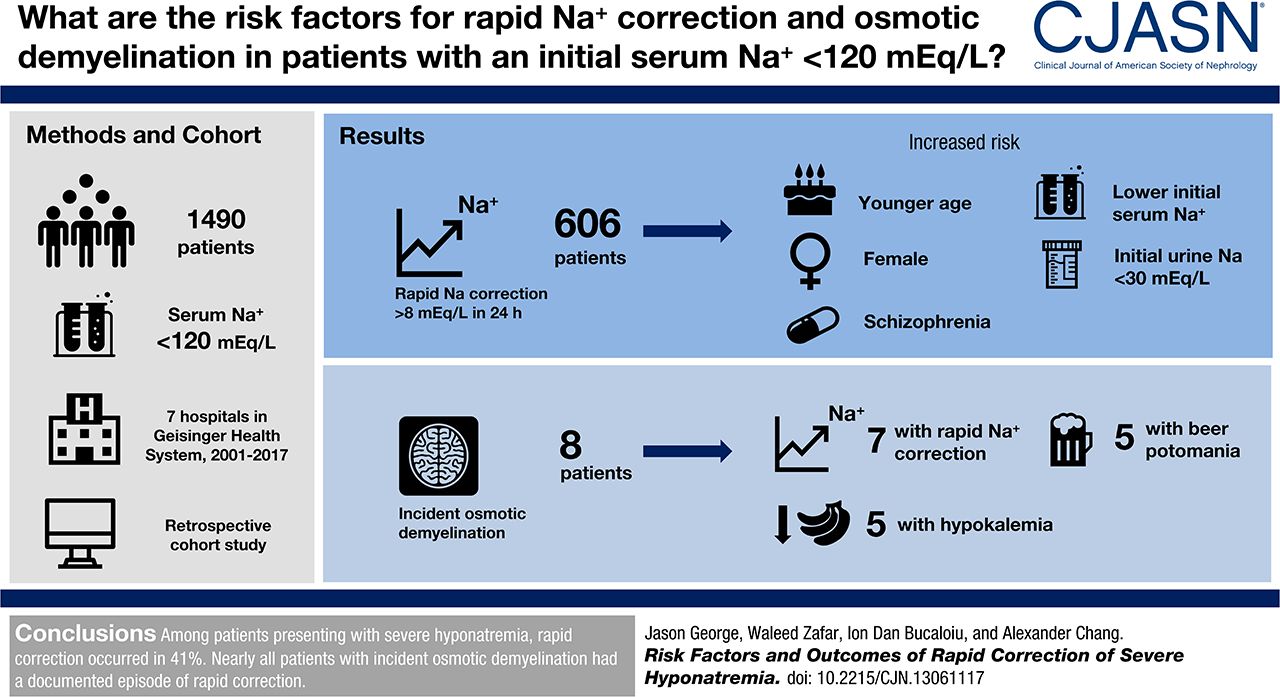
George, Zafar, Bucaloiu, and Chang analyzed the 7 hospitals in the Geisinger Health System to look at the incidence and risk factors for rapid sodium correction and osmotic demyelination.
They included adults with an initial sodium less than 120 mmol/L.
Their primary endpoint was an increase in sodium of more than 8 mmol/L in the first 24 hours.
Looking at the methods, it was not clear to me how they dealt with patients with multiple admissions for hyponatremia. Were they included multiple times? Did they only include the first admission?
They manually looked through every MRI report that was done after the index sodium to look for reports of ODS.
Results
They found 1,490 cases of severe hyponatremia with complete data that matched their inclusion criteria.
Median change in sodium in the first 24 hours was 6.8 mmol/L.
606 (41%) patients broke the speed limit of 8 mmol/L in the first 24 hours.
Risk factors for rapid correction
The authors provided a lot of depth on the risk factors for rapid correction. Here is the unadjusted risk factors.
Demographic:
- Younger (average age of rapid correctors was 63 versus 68 in slow correctors)
- Current smokers (40% of rapid correctors were smokers versus 26% of slow correctors)
- Female gender
Medical history:
- Depression (in 20% of rapid correctors versus 16% of slow correctors)
- Schizophrenia (in 4% of rapid correctors versus 1% of slow correctors)
- No history of hyponatremia (in 59% of rapid correctors versus 73% of slow correctors)
- No history of chronic liver disease (in 6% of rapid correctors versus 8% of slow correctors)
- No history of congestive heart failure (in 12% of rapid correctors versus 19% of slow correctors)
- No history of cancer (in 19% of rapid correctors versus 25% of slow correctors)
Clinical data:
- Lower body mass index (BMI of 26 in rapid correctors versus 28 in slow correctors)
- Lower initial sodium (115 in rapid correctors vs 117 in slow correctors)
- Lower initial urine sodium (35 in rapid correctors vs 43 in slow correctors)
- Lower initial urine potassium (27 in rapid correctors vs 32 in slow correctors)
- Lower initial urine osmolality (270 in rapid correctors vs 369 in slow correctors)
- Have seizures (13% in rapid correctors versus 9% in slow correctors)
They used three different models for multivariate analysis to generate odds ratios for rapid correction of hyponatremia.

Some comments about the risk factors. I suspect the reason that depression actually represents SSRI induced SIADH that rapidly corrects when the drug is withdrawn. The schizophrenia association may represent psychogenic polydipsia, a common manifestation of schizophrenia. Water restriction will result in prompt normalization of the sodium.
Liver, heart, and cancer are all associated with difficult to treat hyponatremia, so no surprise that they saw lower rates of rapid correction among people with those diseases.
The lower BMI and female gender association with rapid correction may be simply due to lower total body water making these patients more susceptible to rapid changes in serum sodium.
Lower urine sodium may indicate patients with volume depletion hyponatremia, a population predisposed to rapid correction of hyponatremia as soon as the volume depletion is corrected, releasing the hypothalamus from the volume stimulated release of ADH and resulting in rapid drop in ADH and rapid, uncontrolled correction of hyponatremia.

The lower urine osmolality likely represents patients with psychogenic polydipsia, tea and toast syndrome, or patients who are already in the midst of spontaneously correcting their sodiums, all patients who will rapidly correct their sodium.
The people who are seizing are a cohort with, presumably, acute symptomatic hyponatremia and rapid correction is less problematic.
Osmotic demyelination syndrome
There were 295 patients who had an MRI after having severe hyponatremia. Nine had radiologic evidence of osmotic demyelination. No patients had an ICD diagnosis of central pontine myelinolysis. One patient had MRI evidence of ODS prior to correction of the sodium. Consistent with medical consensus, nearly all of the patents with incident ODS had rapid correction of hyponatremia:
Of the eight (0.5%) patients who developed incident osmotic demyelination, seven (88%) had documented sodium correction > 8 mEq/L during any 24-hour period before brain MRI.
The one patient with evidence of ODS without rapid correction of hyponatremia had been admitted within a month with a sodium of 105. So it is possible the ODS occurred with that episode rather than the one captured in the study.
The authors listed the characteristics of the patients with ODS:
- Hypovolemia in 75%
- Beer potomania in 63%
- Outpatient thiazide diuretic use in 25%
- Alcohol use disorder 50%
- Malnutrition in 50%
- Hypokalemia in 63%
Having hypovolemia in 75% and beer potomania in 63% means that some patients had to have both of these diagnosis. This portrays a misunderstanding of what constitutes beer potomania. Beer potomania induced hyponatremia is not merely a patient who drinks beer and develops hyponatremia, instead it represents a patient who is unable to clear the water volume in beer due to inadequate solute intake. These patients have maximally dilute urine. On the other hand, patients with volume induced hyponatremia are unable to clear excess water because of hypovolemia-induced ADH release. In one case you have high ADH activity and concentrated urine (volume depletion) and in the other case you have suppression of ADH and dilute urine. There can not be a meaningful combination of the two disorders.
Alcohol use, malnutrition and hypokalemia are all previously recognized risk factors for ODS.
Three patients had received 3% saline for acute neurologic symptoms of hyponatremia. This is critical information because it shows that just because someone has acute manifestations of hyponatremia and an indication for 3%, one must still pay attention to the speed limit. Often acute neurologic symptoms of hyponatremia is thought to indicate a failure to have compensated for chronic hyponatremia and therefore protective against ODS. This is not the case.
The finding that I found most interesting was the clinical outcomes of the patients with ODS.
Five patients with documented osmotic demyelination had recovery with no neurologic deficits, two patients died from unrelated causes, and two were lost to follow-up.
They had 9 cases of ODS and they weren’t able to document even a single case of neurologic devastation. Five of the cases had documented neurologic recovery. This is how Burton Rose describes ODS:

And here is how Sarah and I described it in the fluids book

In my mind if you don’t spasmodic, mirthless laughter you don’t have ODS.
This is consistent with a previous study of dialysis patients with hyponatremia who developed similarly clinically invisible ODS was diagnosed by MRI. From the discussion of Tarhan et al.
Contrary to its classic description, osmotic demyelination syndrome may develop without any presenting signs and symptoms in patients with end-stage renal disease who have undergone recent hemodialysis.
This largely asymptomatic ODS likely explains why not one of the 9 patients had an ICD diagnosis of central pontine myelinolysis (CPM). They did nt have any of the clinical manifestations of CPM.
When I read the abstract and saw that they had 9 cases of ODS in their cohort I jumed out of my shorts. This would be the largest series of DS ever. After reading the study I realize that actually we are in a new era of hyponatrmia where ODS can be devastating (read the three case reports in Brunner et al’s prospective study of MRI evaluation, PDF available on SciHub) or asymptomatic.


 One of the interesting developments in MedTwitter has been the chained tweet to demonstrate a point. I think the master of this is Professor Darrel Francis.
One of the interesting developments in MedTwitter has been the chained tweet to demonstrate a point. I think the master of this is Professor Darrel Francis.












 This article is accompanied by
This article is accompanied by 
